Home
Subscription
July 2026 issue
June 2026 issue
May 2026 issue
April 2026 issue
March 2026 issue
February 2026 issue
January 2026 issue
BACK ISSUES
2025
2024
2023
2022
2021
2020
2019
2018
2017
2016
2015
2014
2013
2012
2011
2010
2009
2008
2007
2006
|
||
|
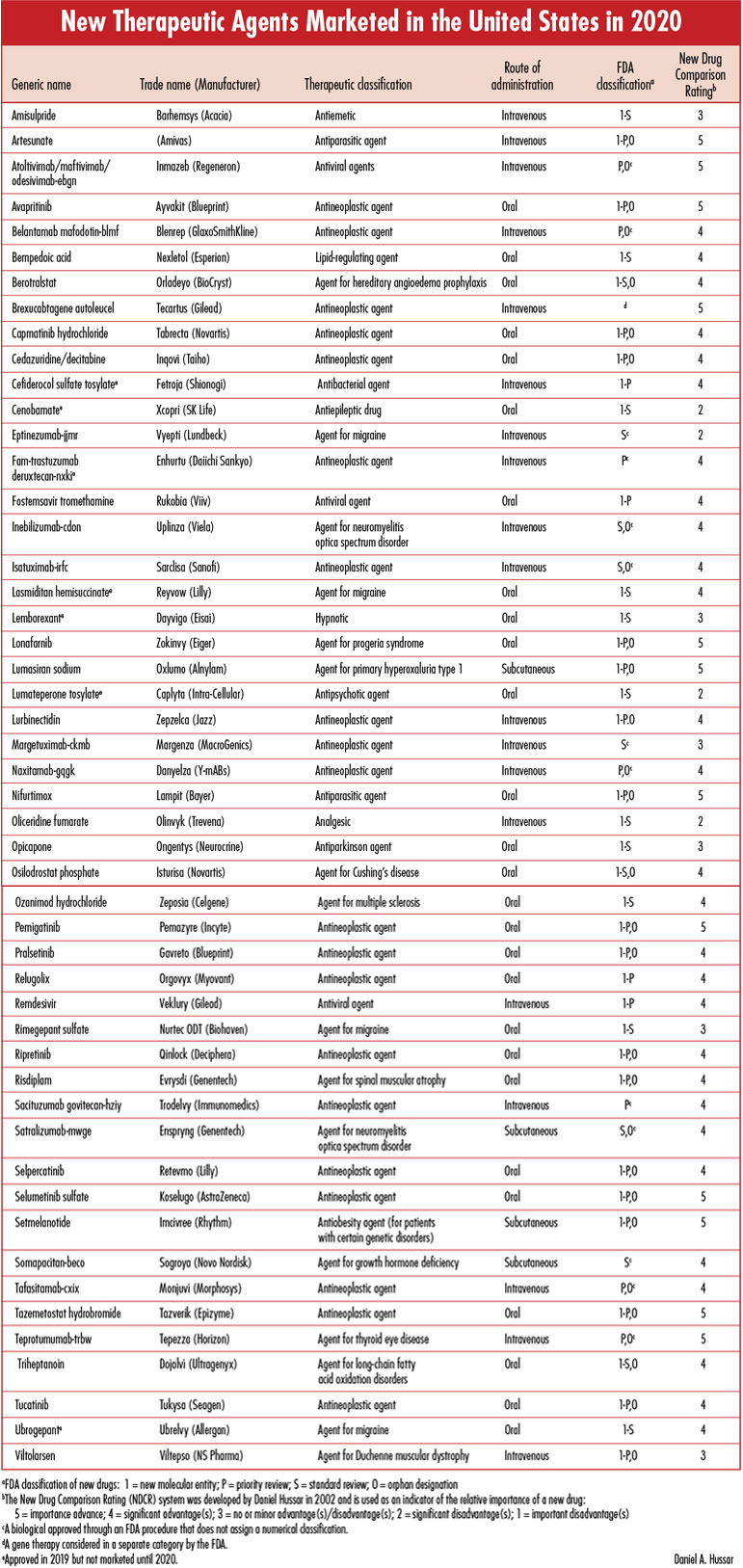
Each issue will include an editorial on a topic that is important for the profession of pharmacy, as well as a review of a new drug that includes a comparison of the new drug with previously marketed drugs that are most similar in activity, and a New Drug Comparison Rating (NDCR) for the new drug. Read on for this month's issue. June 2021 Issue [Download PDF format] In this issue: • Editorial • New Therapeutic Agents Marketed in the United States in 2020 |
||
EDITORIAL:FDA Approval of Aduhelm for Alzheimer's Disease is the Wrong Decision!Very few other medical challenges create the level of fear among patients and their families as a diagnosis or probability of Alzheimer's disease. As one who has written, spoken, and celebrated the development of important new therapeutic agents for more than 50 years, I would be delighted to be describing the benefit and value of a highly effective treatment for this devastating condition. Aducanumab (Aduhelm) is NOT that drug. At best, the FDA decision to approve this drug for the treatment of Alzheimer's disease is greatly premature!The drugs that have been previously approved for the treatment of patients with Alzheimer's disease include the cholinesterase inhibitors such as donepezil (e.g., Aricept) and the N-methyl-D-aspartate (NMDA) receptor antagonist memantine (e.g., Namenda). Unfortunately, the clinical benefit of these agents is very limited. Progress has been made in identifying pathophysiologic features of Alzheimer's disease, although uncertainties and differences of opinion persist among the experts regarding the importance of these characteristics and potential treatments. The identification of accumulated amyloid beta plaques in the brains of patients with Alzheimer's disease has resulted in investigations of agents that reduce the number of these plaques. However, to date, studies of these investigational agents have failed to demonstrate clinical effectiveness, with the consequence that the studies have been discontinued. AducanumabAducanumab-avwa is a human monoclonal antibody directed against forms of amyloid beta. It was evaluated by Biogen in two placebo-controlled studies in patients with confirmed presence of amyloid pathology and mild cognitive impairment or mild dementia. There is agreement that the drug consistently reduces amyloid beta plaques in the brain in a dose- and time-dependent manner. The reduction in this surrogate marker is thought to predict clinical benefit but is not itself a measure of clinical benefit.In both studies patients were randomized to receive aducanumab low dose, aducanumab high dose, or placebo intravenously every 4 weeks for 18 months. At a point during the trials a "futility analysis" was conducted that appeared to indicate that aducanumab was not likely to be more effective than placebo and, in March 2019, Biogen terminated both trials prior to their planned completion. In the context of previous clinical trial failures of other amyloid-targeted investigational agents, the failure of yet another agent thought to have potential benefit was very disappointing but not shocking. It is reasonable to think that, if there was any possibility that these studies of aducanumab would show clinical benefit in the treatment of a devastating disease, and also have the potential for many billions of dollars of revenue, Biogen would not have terminated the studies early. But this was only the first of a series of controversial decisions. Following early termination of the clinical trials, Biogen continued to review and reanalyze the available data/results. In one of the clinical trials, no statistically significant differences on the primary efficacy endpoint were observed between the aducanumab-treated (high dose or low dose) and placebo-treated patients. However, in the other clinical trial the high dose (but not the low dose) of aducanumab was thought to reduce clinical decline, as reflected by a statistically significant treatment effect on the primary and secondary efficacy endpoints compared to placebo. Notwithstanding the failure to identify clinical benefit in one of the studies, Biogen announced in November 2019 that it would use information from its additional analysis of data from the other uncompleted study to support the development of a biological license application (BLA) to submit to the FDA. Following receipt of Biogen's application for approval of aducanumab, the FDA convened its Peripheral and Central Nervous Systems Drugs Advisory Committee to review the information submitted and provide recommendations. The Committee met in November 2020 and voted overwhelmingly that the information reviewed did not justify approval of the drug. Although FDA officials are not required to make decisions that are consistent with the recommendations of its Advisory Committees, they usually do so and the resultant expectation that the application for approval would be rejected prompted intense publicity and lobbying in support of approval of aducanumab from patients and advocacy groups, as well as those with financial and political interests. On June 7, 2021 the FDA announced that it approved aducanumab under the provisions of the accelerated approval process that is based on a drug's effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit for patients. Because the FDA's action was not based on the demonstration of clinical benefit, it also required Biogen to conduct a post-approval trial to verify that the drug provides the expected clinical benefit. Those who support the FDA's decision say that it provides hope for and perhaps actual clinical benefit, greater and more timely access to treatment for patients with Alzheimer's disease, as well as incentives for pharmaceutical companies and investors to expand research efforts that will result in more medications that will provide effective treatment. Critics of the decision, including this writer, contend that a clinical benefit has not been clearly demonstrated, that the FDA should have required completion of another clinical trial to verify a clinical benefit before approving the drug, that the earlier failures of multiple amyloid-targeted investigational agents to have demonstrated clinical benefit do not inspire confidence that the experience with aducanumab will be any different, that the FDA has reduced its standards for approving new drugs and also reduced the credibility of the approval process and the role of Advisory Committees, and that ready access to aducanumab will increase the difficulty in recruiting patients, who might be assigned to a placebo group, to participate in clinical trials of potential effective treatments for Alzheimer's disease. Soon after the FDA announcement, three members resigned from the Advisory Committee that had recommended against approval, with one stating that the decision was "probably the worst drug approval decision in recent U.S. history," and another indicating that the decision would undermine public trust and medical innovation. The recommended maintenance dosage of aducanumab is 10 mg/kg administered by intravenous infusion every 4 weeks, and the anticipated cost of the medication for one year would be approximately $56,000 for each patient. An analysis conducted by the Kaiser Family Foundation estimates that if 500,000 Medicare recipients (of the approximately 6.2 million Americans with Alzheimer's disease) are prescribed aducanumab, it would cost the Medicare program nearly $29 billion a year, much more than any other medication. Some patients who are treated with the drug could have copayments of about $11,500 annually, and Medicare premiums are likely to be increased. The FDA has the authority to approve a medication based on improvement in a surrogate marker, but I consider it unwise to reject the almost unanimous recommendation of its Advisory Committee in taking the action it did. The FDA has stated that failure of aducanumab to meet the efficacy endpoint in the post-approval trial it has required Biogen to conduct could result in removal of the drug from the market. However, I can't imagine that would happen because, even if clinical benefit is not demonstrated in the relatively small controlled clinical trial, the uncontrolled experience in the much larger number of patients for whom it can now be prescribed will become a de facto study in which some patients, caregivers, and prescribers will insist that benefit has been provided. The opposition to removing the drug from the market will be far greater than the support for approving it now. For a drug for which the FDA provides accelerated approval based on improvement in a surrogate marker, and not clinical benefit, the experience of the patients for whom it is prescribed will be an important factor in the decision as to whether the drug will be permitted to remain on the market. A pharmaceutical company should make such a drug available without charge until the clinical benefit is demonstrated. Daniel A. Hussar
danandsue3@verizon.net |
||
| ||